Resposables y recursos del proyecto
- Reponsables académicos:
- Uri O. García Vázquez
- Marysol Trujano Ortega
- Asesor GridUNAM:
- Leobardo Itehua Rico
Introducción
Este proyecto aborda diferentes aspectos de la estructura genética poblacional, límites de especies, sus relaciones filogenéticas y la historia biogeográfica de tres grupos de organismos considerados modelos biológicos (reptiles, anfibios y mariposas) por la capacidad informativa de sus patrones de diversidad, con la finalidad de proveer información acerca de las causas ecológicas e históricas de dichos patrones y con ello la comprensión del origen y evolución de ambientes que presenta tasas altas de modificación de México y América Central y del Sur.
Descripción del trabajo a realizar
Se obtuvieron diferentes conjuntos de datos genómicos obtenidos mediante las técnicas de secuenciación masiva de DNA ddRAdSeq y 3RAdSeq que suman 581 muestras de reptiles, anfibios y mariposas que representan cuatro géneros Rhadinaea (61 muestras), Rheohyla (90 muestras), Plesioarida (250 muestras) y Emesis (180 muestras) distribuidos en América y con una relación estrecha con los ambientes que habitan (Fig. 1). Estos datos se secuenciaron en la plataforma Illumina Hi-seqX y poseen cerca de 30-40 mil loci.
Edición y alineamiento.
Los datos se editarán y recuperarán con el pipeline ipyRAD (Eaton y Overcast 2020). Se determinarán los umbrales de agrupación de similitudes de secuencia para las lecturas filtradas de acuerdo con McCartney et al. (2019) (Fig.2).
Análisis de datos moleculares.
El conjunto de datos se analizará en PartitionFinder (Lanfear et al. 2012). Se realizarán análisis de máxima verosimilitud (ML) y bayesianos (IB). Se utilizará RAxML (Stamatakis 2006) para las reconstrucciones de ML con el modelo GTRCAT y 100 réplicas de bootstrap no paramétrico para evaluar el soporte de los nodos. El análisis IB se realizará en MrBayes (Ronquist et al. 2012). Se estimará la probabilidad posterior de cada clado (Fig. 3).
Límites de especie.
Se examinará la estructura de las poblaciones utilizando algoritmos de agrupamiento genético implementados en Structure (Pritchard et al. 2000) y PhyloNetWork (Solís-Lemus et al. 2017). Se realizará un análisis de BPP para realizar conjuntamente la delimitación de especies y la inferencia de árboles de especies bajo el modelo coalescente de especies múltiples, se realizarán varios análisis dividiendo grupos de poblaciones como lo sugieren los análisis poblacionales (Fig. 4).
Análisis morfológico.
Los taxones nuevos se describirán, ilustrarán y compararán en un contexto filogenético. Diversos caracteres se explorarán en una resolución fina con herramientas de microscopía estereoscópica (ME) y electrónica de barrido (MEB). Las técnicas empleadas serán diafanización, tinción, digestión y rehidratación.
Estimación de tiempos de divergencia.
Se empleará un enfoque bayesiano con un modelo de reloj molecular relajado en BEAST2 (Bouckaert et al. 2014). Se calibrará con diversos fósiles de acuerdo con el grupo de estudio. Los tiempos de divergencia obtenidos se compararán con diversas hipótesis geológicas (Fig. 5).
Análisis biogeográfico.
Se reconstruirán las áreas ancestrales de cada especie por medio de un análisis bayesiano binario implementado en el programa RASP (Yu et al. 2015). Se utilizarán todos los árboles obtenidos en BEAST. Cada uno de los taxones incluidos se codificará de acuerdo con su procedencia geográfica y de acuerdo con las regionalizaciones bióticas de América (Fig. 6).
Cálculos y Software a utilizar
-
Alineamiento de secuencias con diferentes conjuntos de datos (ipyrad). En un job con una muestra de 61 secuencias (7.3 GB); 6 cores y 16 GB RAM, se estimó un tiempo de 5:23 hrs. con archivos de salida de 10 GB. Se requieren 10 repeticiones modificando valores de parámetro (clust_threshold 0.85-0.95). Tiempo total estimado de 2.2 días con 100GB de almacenamiento. Este análisis se realizara por duplicado, con un numero reducido de muestras.
-
Estructura poblacional con dos análisis (conStruct implementado en R y Phylonetwork en Julia). En uno de los análisis con archivo de entrada de 27 muestras (1 GB); 4 cores y 8 GB RAM, se estima un tiempo de dos días; mientras que el otro análisis requiere el archivo de muestras y un árbol filogenético (2 GB); 10 cores y 32 GB RAM, se estimó tiempo de cinco días.
-
Reconstrucción filogenética (RaxML y Mr.Bayes). Archivo de entrada con 61 muestras (1 GB), se estima un Job con 128 cores y 64 GB RAM, tarda 0.5 hrs. En el caso de los análisis bayesianos se estima el triple de tiempo.
-
Divergencia y Demografía (BPP). Archivo de entrada con 27 muestras (3 GB); 10 cores y 32 GB RAM, se estiman cuatro días de análisis.
Figuras tomadas de artículos propios


Figura 1. Modelo biológico y su distribución geográfica.

Figura 2. Métricas del alineamiento.

Figura 3. Recostrucción filogenética.

Figura 4. Análisis poblacional, red de haplotipos.

Figura 5. Análisis de divergencia.
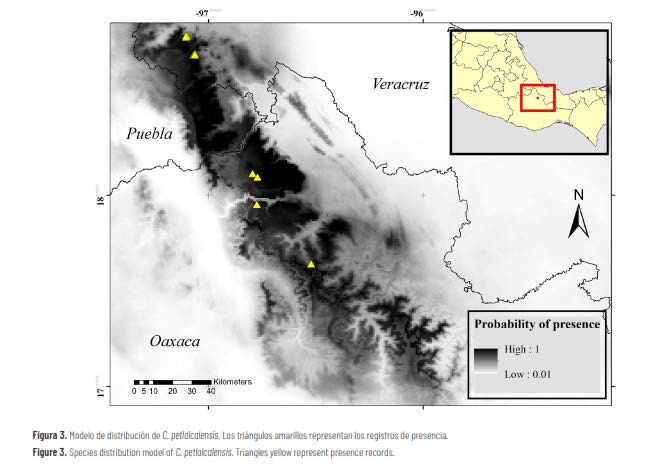
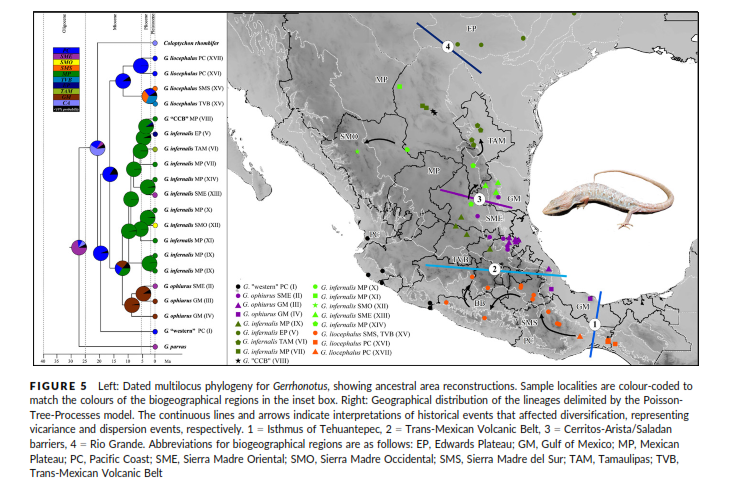
Figura 6. Análisis ecológico y biogeográfico.
Referenias
Software
Estrategia para uso en la GridUNAM
Por la cantidad y diversidad de software a utilizar, se empleará un contenedor.